Medium Chain Acyl CoA Dehydrogenase Deficiency (MCADD) OMIM # 201450
Edited by: Dianne M. Frazier, PhD, MPH, RD
Last Updated: February 10, 2008
Background
Definition: Medium chain acyl CoA dehydrogenase deficiency (MCADD) is caused by mutations in the medium chain acyl CoA dehydrogenase (MCAD) gene leading to insufficient enzymatic activity to allow complete mitochondrial beta oxidation of fatty acids. Further description of MCADD.
Incidence: In the US, the incidence is ~ 1 in 10,000 births among Caucasians. There is a lower incidence among African Americans and Latinos (1)
Genetics: MCADD is inherited as an autosomal recessive disorder. The gene is found on 1p31 and has 12 exons (2,3). The most common mutation, among those of Northern European descent , is 985A>G ( K329E).
Pre-symptomatic detection: It is possible to detect MCADD through tandem mass spectrometry (4) newborn screening of blood spots. Although the definition of a positive screen is established by each state's program, the minimum requirement is that the medium chain (C8 and and possibly also C6,C10 and C10:1) acyl carnitines be elevated and the C8 acylcarnitine be greater than the C10.
Confirmatory testing: For any positive screen, further confirmatory testing is necessary before a diagnosis of MCADD can be made. This confirmatory testing usually includes a plasma acylcarnitine profile, urine organic acids, DNA mutation analysis and plasma carnitine. A repeat newborn screen, in and of itself, is not sufficient for confirmatory testing for MCADD. See American College of Medical Genetics guidelines.
Note: infants on medium chain triglyceride (MCT)- containing formula or breast milk enhancers ( see MCT in infant formulas) may have MS/MS screening results indistinguishable from those of a newborn with true MCADD.
Signs and Symptoms
Any individual with MCADD whose energy needs are not being met from exogenous sources, and who has to rely on stored fat, runs the risk of metabolic decompensation. MCADD infants and young children, especially during intercurrent illness, are at greater risk than older children and adults. Symptoms can range from fatigue, lethargy, hypotonia, and progress to vomiting, acidosis, hyperammonemia, hypoketotic hypoglycemia, fatty infiltration of the liver, coma and death (5). A MCADD infant, who is healthy and feeding normally, will show no signs or symptoms of the disorder.
back to top
Laboratory Findings
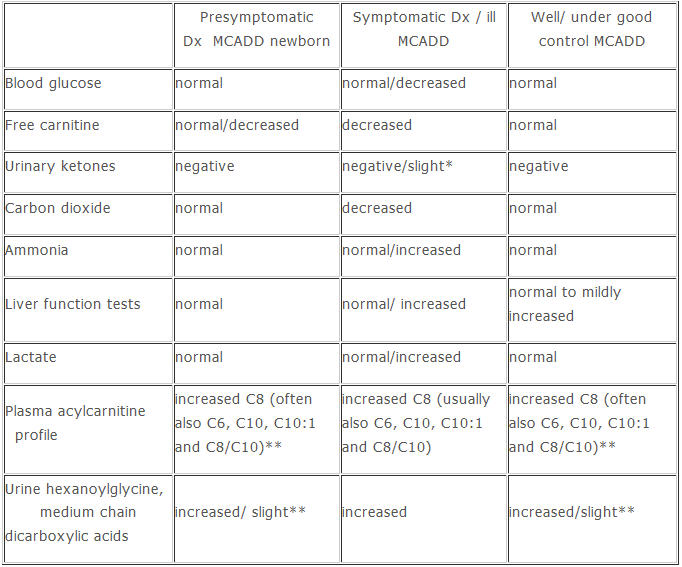
* Slight ketones may be present in fasting MCADD individuals with some residual MCAD activity
** Elevations of the urinary dicarboxylic acids and acyl plasma carnitines may be less pronounced in MCADD individuals with some residual MCAD activity
back to top
Biochemical Basis of the Disorder
Role of glucose: Glucose is the preferred energy source in cells, and glucose derived from stored glycogen is used during short term fasting. Stored fat is utilized for energy when glucose sources are being depleted, whether that is due to inadequate intake or extraordinary energy requirements.
Oxidation of stored fats: Fats are stored as the long chain species (C16, C18) and are oxidized by chain length -specific enzymes. The long chain fatty acids are oxidized in two carbon units by a series of four long chain-specific enzymes. As oxidation progresses, and the chain length is reduced to 8 -10 carbons, the medium chain length-specific oxidative enzymes are utilized (6).
Loss of MCAD activity: Deleterious mutations of the MCAD gene result in a protein without sufficient enzyme activity to catalyze the first step of oxidation of medium chain length fatty acids. The rate of oxidation slows and this prevents the cells from generating maximum energy from stored fatty acids. This limits the production of ketones which are usually seen during fasting in individuals when fatty acids can be completely oxidized to acetyl CoA. In MCADD, the amount of energy normally derived from ketones during a fast is limited.
Physiological effects of increased acyl CoA intermediates: When the medium chain fatty acids cannot be further oxidized, they accumulate as the medium chain CoA intermediates. If there is unsufficuent carnitine available to exchange for the CoA moiety, and help transport the accumulated intermediates out of the mitochondria, CoA may become a limiting factor. Other catabolic pathways which utilize CoA may be unable to function to produce energy.
back to top
Chronic Management
Before MS/MS newborn screening, nearly all cases of MCADD were diagnosed symptomatically. Intervention and treatment often addressed the serious consequences of metabolic decompensation. As more presymtomatic infants have been identified, treatments have become directed toward prevention of metabolic decompensation. There is still controversy regarding the stringency of intervention and monitoring. The GMDI guidelines are based on a review of the literature, presentations at professional meetings, personal communications, and established protocols at several large metabolic centers(7,8). GMDI expects that as more data accumulates through clinical experience and peer-reviewed research, the guidelines may be revised. You are invited to add your experience and comments by clicking on the "Comments" section at the end of this protocol.
Goals: The primary intervention goal for MCADD individuals is to avoid situations in which the cells must rely soley on stored fats for energy (i.e., avoid fasting).
Precautions: Acute illness is often the cause of metabolic decompensation due to poor intake and increased energy demands.
Emergency letter: Families should be provided with an emergency protocol/letter that can be carried with them at all times. They should be strongly advised to seek medical attention if the individual with MCADD has an acute illness accompanied by poor intake, vomiting and/or lethargy. The letter should contain: patient identifiers, description of the disorder, emergency treatment protocol, and contact information for the metabolic specialist.
Medical alert tag: A medical alert tag may be helpful when children are no longer under constant surveillence by their caregivers (i.e, in school or traveling). Minimially, the information on the tag should include: "MCADD", "at risk for hypoglycemia", "give glucose".
Glucose source: A concentrated glucose source should be available at all times to be used to treat hypoglycemia until emergency care can be accessed. Possible sources are: Instaglucose, liquid polycose and gel cake frosting.
Frequency of feedings: The frequency of feeding for an MCADD infant (9), who is well, should be no different than that for a non-MCADD infant. A rule of thumb: maximum time of nighttime 'fasting" between birth and 4 months of age should not exceed 4 hours; between 5 and 12 months, an additional hour can be added for each month. Some clinics recommend checking an infant's blood glucose levels before feeding in the morning to determine tolerance to nighttime fasting and to individualize fasting recommendations, if necessary. In childhood, age-appropriate meal and snack schedules should be followed. All MCADD individuals should avoid skipping meals and weight loss diets which recommend fasting. Prolonged and/or intense exercise should be "covered" with adequate carbohydrate intake and hydration. I.V. glucose is recommended whenever a medical procedure requires fasting (NPO) for several hours before and after the procedure.
Diet composition in infancy: Regular infant formula or breast milk can be continued in MCADD infants. Typical infant formulas and breast milk can contain up to 55% of the Kcal as fat, much of which is used for anabolic processes during the first year of life. If an infant is breast feeding, it is important to monitor adequate breast milk production by assessing the infant's weight gain and satiety. Formulas (and breast milk enhancers) containing MCT oil are not appropriate for the MCADD infant. See MCT in infant formulas
Diet composition in children and adults: Heart healthy diets with appropriate Kcal for age and size are recommended after infancy. Nutritional intervention may be needed to counsel individuals about meal planning so that they get approximately 30% of Kcal from fat, include fruits and vegetables on a daily basis, utilize complex carbohydrates, and avoid overfeeding or exceeding their kcal needs. The long term benefits of appropriate weight gain, the health consequences of obesity, and the danger of fasting for an MCADD individual should be emphasized. The popular very high protein/fat and low carbohydrate diets are not appropriate.
Supplementation: The dietary recommendations for MCADD do not include restriction of any food group or single nutrient, so there is no a priori need for nutrient supplementation.
Carnitine: L-carnitine, which is synthesized endogenously, may become relatively depleted in MCADD as it remains conjugated to the accumulated medium chain fatty acids and is excreted as the acylcarnitine. Recommended dosages for infants are 50-100 mg/kg. The need for, and the amount of, supplemetation in children and adults is determined by monitoring plasma free carnitine concentration according to the individual clinic's protocols. A plasma free carnitine in the 25-35 micromolar range is a usual goal.
Glucose: A few clinics use cornstarch, Polycose or other high carbohydrate supplements/snacks as a glucose source for MCADD infants and children before the nighttime fast. Unless these individuals are unwell and/or eating less than normal, glucose supplementation should not be necessary.
back to top
Monitoring
Anthropometrics: MCADD infants and children should follow a normal growth curve pattern for age and sex. Guidance should be given to parents who are overfeeding because they fear fasting-induced hypoglycemia, or are underfeeding because they are over-restricting dietary fats.
Glucose testing: There is no need to do frequent blood glucose measurements in MCADD individuals. Some clinics instruct parents to measure blood glucose levels at the end of the longest recommended sleeping interval to determine if the infant needs to be awakened earlier for a feeding. These measurements can also provide a baseline glucose measurement for an individuaI that can be helpful when evaluating glucose measurements during illness. Some clinics also recommend checking a glucose level when the infant or young child seems unusually sleepy, is difficult to awaken or is uninterested in eating, but without any other signs of illness. If home glucose monitoring is desired, a glucometer sensitive to low blood glucose concentrations should be chosen.
Labratory testing: After the initial confirmatory testing has been done, the only routine laboratory measurement needed is blood carntine which gives total carnitine, free carnitine and acyl carnitine. Individuals who are on supplemental carnitine should have periodic assessment of their free carnitine. There is no evidence to support the necessity of repeating either the acyl carnitine profile or the urine organic acid analysis.
Dietary assessment: Diet records should be assessed at each clinic visit. The factors to assess include: adherence to the dietary guidelines for MCADD (above); appropriate spacing of meals and snacks; and approriate nutrient intake for age and weight.
back to top
Acute Management
For the individual with MCADD, intervention during illness is crucial. Illness increases caloric demands, while at the same time poor appetite and/or vomiting and diarrhea may limit intake. The intervention strategies for illness should be the most emphasized aspect of counseling for the MCADD individual and his caretakers.
At home care: At home care for an ill MCADD individual is possible only if fasting can be avoided by providing adequate oral carbohydrate and fluid. Although rehydration fluids such as Pedialyte are extremely useful for ill infants and children without MCADD, the solution, when used exclusively, does not contain adequate glucose to maintain blood glucose levels in an ill MCADD infant or child. Sweetened fluids containing approximately 1/2 tsp sugar per ounce should be able to maintain blood glucose if given in quantities to meet the fluid requirement. Fluids and/or meals should be small and frequent. Ill infants and children should be awakened during the night for feeding and assessing their condition. Carbohydrate-containing oral rehydration may allow return to regular oral intake.
Emergency care: It is imperative that caregivers understand the importance of immediate and decisive action when the MCADD individual:
Is ill and not capable of sufficient oral intake, and/ or
shows signs of lethargy, hypotonia, decline in mental status, and/or
has blood glucose levels < 60mg/dl, which cannot be improved with oral feeding.
NOTE: Low blood glucose levels may not develop until later in an illness. It should be
emphasized that emergency care must be initiated if symptoms are present.
Caregivers should not rely solely on the blood glucose levels to initiate care.
Oral glucose: The use of a concentrated oral glucose source ( see above) may be lifesaving while awaiting emergency care. This can be rubbed, at frequent intervals, on the inside of the cheeks and under the tongue, even if the individual is non-responsive.
Emergency protocol letter: A copy of the emergency letter (see above) should be given to the EMS or ED personnel.
IV treatment: 10% dextrose, with appropriate electrolytes run at 1.5 times maintenance fluid rate, is recommended to establish and maintain normaglycemia.
IV carnitine: When oral intake of carnitine is decreased and/or carnitine requirements increased due to catabolism, IV carnitine at a dose of 100mg/kg body weight may be needed for the short term. Because not all hospitals have IV carnitine available, it may be possible to continue to give liquid carnitine orally since only small volumes are typically needed. Many recommend doubling the oral carnitine dosage during illness.
Length of stay: Discharge from the medical facility should not occur until the MCADD individual is able to consume sufficient oral intake to maintain normaglycemia.
back to top
Special Circumstances
Surgery: Fasting before surgical procedures and during recovery puts an MCADD individual at risk. Most agree that clear liquids can be given up to 4 hours before surgery, but IV glucose should be started before the procedure and continued until the patient can consume adequate oral intake and can maintain normoglycemia. Post -surgical pain and nausea may affect appetite and intake.
Trauma: Trauma and injury increase caloric needs, but pain and possible alteration in mental status may decrease intake. Careful monitoring is very important. IV glucose should be used whenever there is any concern about catabolism.
Pregnancy and child birth in an MCADD female: During pregnancy, plasma free carnitine levels will decline(10). Dosages of 100mg/kg are recommended, but free carnitine may still not reach pre-pregnancy levels. IV glucose should be started as soon as labor begins and continued until the patient has adequate oral intake and can maintain normoglycemia.
Neonatal care for an MCADD sibling: Families, who already have a child/children with MCADD, have a 25% risk of reccurence of MCADD with each pregnancy. If they do not have prenatal diagnosis, it may take several days to have either screening or diagnostic testing results available. It is important to monitor this at-risk infant closely. If the mom wishes to breast feed, glucose water or small amounts of formula, given after the newborn has been been put to the breast, can help prevent catabolism until breast milk production is adequate.
back to top
Education
Learning Objectives: The individual with MCADD and/or the caregiver will:
Understand importance of avoiding fasting and eating meals on a regular schedule
Understand the need for maintaining appropriate blood glucose levels
Understand the emergency protocol and when to use it
Recognize signs of illness
Offer foods/formulas with appropriate type and amount of fat
Recognize the genetic basis of MCADD
Review information and expand knowledge at each encounter
Educational Resources:
Star-G
Fatty Acid Oxidation Support Group
back to top
References
1. Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH, Muenzer J (2006). The tandem mass spectrometry newborn screening experience in North Carolina: 1997-2005. J Inhert Metab Dis 29: 76-85.
2. Andresen BS, Bross P, Jensen TG, Knudsen I, Winter V, Kolvraa S, Bolund L, Gregersen N (1995). Molecular diagnosis and characterization of medium-chain acyl-CoA dehydrogenase deficiency. Scand J Clin Lab Invest Suppl. 220: 9-25
3. Andresen BS, Jensen TG, Bross P, Knudsen I, Winter V, Kolvraa S, Bolund L, Ding JH, Chen YT, Van Hove JL, et al. (1994). Disease-causing mutations in exon 11 of the medium-chain acyl-CoA dehydrogenase gene. Am J Hum Genet. 54(5): 975-66
4. Van Hove JL, Zhang W, Kahler SG, Roe CR, Chen YT, Tereda N, Chace DH, Iafolla AK, Ding JH, Millington DS (1993). Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. Am J Hum Genet. 52: 958-66
5. Iafolla AK, Thompson Jr RJ, Roe CR (1994). Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediat. 124: 409-415
6. Saudabray JM, Martin D, deLonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Bianey-Saban C, Bonnefont JP, Tabier D, Kamoun P, Brivet MR (1999). Recognition and management of fatty acid oxidation defets: A series of 106 patients. J Inher Metab Dis 22: 488-502
7. Solis JO, Singh RH (2002) Management of fatty acid oxidation disorders: a survey of current treatment strategies. J Am Diet Assoc 102: 1800-1803
8. Wilcken B, Haas M, Joy P, Wiley V, Chaplin M, Black C, Fletcher J, McGill J, Boneh A (2007) Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet 369(9555): 37-42
9. Derks TG, van Spronsen FJ, Rake JP, van der Hilst CS, Span MM, Smit GP (2007) Safe and unsafe duration of fasting for children with MCAD deficiency. Eur J Pediatr 166(1): 5-11
10. Koumantakis E, Sifakis S, Koumantaki Y, Hassan E, Matalliotakis I, Papadopoulou E, Evangeliou A (2001) Plasma carnitine levels of pregnant adolescents in labor. J Pediatr Adolesc Gynecol 14(2): 65-9
back to top